











































*************************************
Complement deficiency
Deficiencies of the complement components have been reported for most of the constituents.
These deficiencies can be inherited or acquired and complete or partial.
Complement deficiency and disease association includes the following:
-
Deficiency of early components of classical pathway (C1, C4, C2): Autoimmune disease, especially systemic lupus erythematosus (SLE), is the most common presentation in patients with early component deficiency.
The incidence rates of SLE in individuals with C1q and C4 are reported to be around 90% and 75%, respectively. Patients with C2 deficiency develop SLE with lesser frequency (around 15%). The proposed mechanisms of high incidence of autoimmune diseases include impaired clearance of immune complex and apoptotic cells and loss of complement-dependent B-cell tolerance. Recurrent bacterial infection is common in patients with C2 deficiency. Atherosclerosis also appears to occur at a higher frequency in individuals with C2 deficiency.
-
MBL pathway
- MBL deficiency is linked with frequent pyogenic infection, including pneumococcal infection in infants and young children. Severe pneumococcal disease is also reported in patients with MASP-2 deficiency.
- In addition, MBL deficiency is 2-3 times as common in patients with SLE as in the general population. Others also report an increase in cardiovascular diseases associated with atherosclerosis in patients with MBL deficiency.
- Schizophrenia is a severe mental disorder, with worldwide prevalence of 1–1.5%. Immunological research in schizophrenia indicates that infectious or autoimmune processes might play a role in the etiopathogenesis.
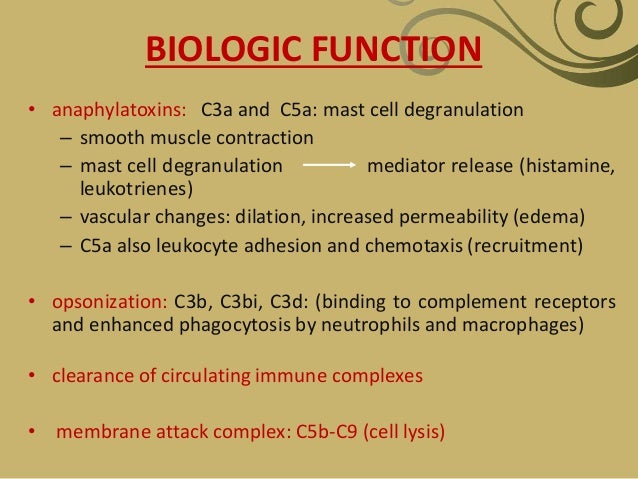
- The complement system is a major mediator of innate immune defense against infection and contributes to many functions of the immune system including inflammation, opsonization and cell lysis.
- MBL activates the complement system via the lectin pathway.
- Inherited MBL deficiency, common in most human populations, predisposes to infectious and autoimmune diseases.
- A modulatory role for MBL in autoimmune disease has been reported. Investigations in Denmark and Hong Kong have suggested that MBL variant alleles are associated with both severity and early onset of disease in patients with rheumatoid arthritis. [3] However, patients with late-onset disease who were also homozygous for wild-type MBL genes were more likely to show evidence of persistent inflammation.
-
Alternative pathway (properdin, factor B, factor D): This is associated with severe fulminant neisserial infections with a high mortality rate.
-
C3, factor H, and factor I: Deficiency of these factors predisposes individuals to severe pyogenic bacterial infections. Factor H and factor I deficiencies cause secondary C3 deficiency with C3 consumption and impose the same infectious risk as primary C3 deficiency. Factor H deficiency is also associated with atypical (diarrhea-negative) hemolytic-uremic syndrome (HUS) and glomerulonephritis. C3 deficiency is associated with membranoproliferative glomerulonephritis.
-
Terminal pathway (C5-C9): The lack of MAC formation results in severe recurrent infection by Neisseria gonorrhoeae or Neisseria meningitidis.
-
C1INH: Primary and secondary deficiency of C1INH leads to uninhibited cleavage of C4 by C1s. Hereditary angioedema (HAE) is caused by heterozygous deficiency of C1INH and characterized by recurrent episodes of angioedema, which usually subside within 48-72 hours. C1INH is a regulator for C1r/C1s, as well as for Hageman factor, clotting factor XI, plasma kallikrein, and plasmin. Angioedema is caused by various mediators that involve these pathways.
-
DAF and CD59:Defects of these 2 regulatory proteins make erythrocytes highly susceptible to complement-mediated cell lysis. This is typically seen in patients with paroxysmal nocturnal hemoglobinuria (PNH), which has clinical features that are characterized by hemoglobinuria and venous thrombosis of major vessels.
************************************************************************
Complement Deficiency Clinical Presentation
Physical
See the list below:
-
Classical pathway deficiencies – C1, C4, and C2
- C1 deficiency generally leads to severe immune complex disease with features of SLE and glomerulonephritis. Serum levels of anti-C1q antibodies were increased significantly in patients with SLE and proliferative nephritis. Among different serological parameters for assessing the renal activity of SLE, only anti-C1q antibodies titers showed significant differences between quiescent and active lupus nephritis. Patients with C2 deficiency are reported to reveal anti-C1q antibody in the absence of anti-dsDNA.
- Complete C4 deficiency is rare. Two genes, C4A and C4B, encode the C4 complement. Both genes are highly polymorphic, and, to date, at least 35 different alleles have been described. Almost all patients with complete C4 deficiency have discoid or SLE, with or without associated glomerulonephritis.
- C2 deficiency has been the most commonly reported classical pathway defect. Skin and joint manifestations are common, and renal disease is relatively rare. Patients with C2 deficiency are also reported to have recurrent or invasive infections.
Deficiencies of the classical complement pathway have been strongly linked to the development of autoimmune disorders, especially those in which excessive immune complexes are formed. Patients may develop collagen vascular disorders, mainly SLE. Ninety-five percent of patients with C1q deficiency, 75% of those with C4 deficiency, 30% of those who are deficient in C2, and 50-55% of those who are deficient in C1r and C1s (deficiency usually involves both C1r and C1s) develop collagen vascular diseases (mainly SLE). Evidence suggests that this may result from the combination of impaired complement-dependent B-cell tolerance and impaired clearance of immune complex and apoptotic cells.
-
C3 deficiency
- C3 deficiency is rare. Because of its importance as a convergence point of the 3 complement pathways, all patients with C3 deficiency develop recurrent, severe, pyrogenic infections early in life. Some patients may also develop membranoproliferative glomerulonephritis.
- C3 deficiency leads to an inability to formulate membrane attack complex (MAC), thus markedly compromising chemotactic and bactericidal activities of the complement cascade.
- Patients with C3 deficiency generally have less than 1% of the normal amount of C3 antigen and C3 function in their serum.
- Because of ineffective opsonization of pathogens, patients with C3 deficiency present with severe recurrent pyogenic infections, mainly caused by meningococci and pneumococci.
- According to published studies, 78% of patients have repeated infections, and 79% experience autoimmune disorders, such as arthralgia and vasculitic rashes, lupuslike syndrome, and membranoproliferative glomerulonephritis.
-
Alternate pathway deficiencies (properdin, factor B, factor D)
- These deficiencies are rare. Properdin deficiency is associated with increased risk of infections with encapsulated bacterial organisms. Patients often present with a history of invasive meningococcal diseases, as well as severe pneumococcal and H influenzae infection. Recurrent infections may be rare in patients with properdin deficiency because the classical pathway can take over the actions of the alternate pathway once antigen-specific adaptive immune responses are established with immunoglobulin (Ig)G and IgM antibody formation.
- Only 3 adults with factor D deficiency have been described in the literature. A 6-day-old newborn boy with pneumococcal sepsis and meningitis was diagnosed with complete deficiency of factor D and diminished functional factor B. The addition of purified factor D restored the capacity of the patient’s serum to generate the alternate pathway fluid-phase C3 convertase (C3bBbP) in response to zymosan. This also restored activity of factor B in the patient’s serum. In contrast, the addition of factor B did not alter factor D activity in the serum, indicating requirement of factor D for function of factor B.
-
Late complement component deficiencies – C5, C6, C7, C8, and C9
- MAC cannot be formed; hence, bactericidal activity is depressed.
- Patients are susceptible to recurrent pyogenic infections.
- Patients typically present with meningococcal meningitis or extragenital gonococcal infection.
- Two thirds of patients experience at least one episode of meningococcal disease, and as many as one half of patients experience recurrent neisserial infections.
- Patients with C9 deficiency have the ability to kill Neisseria, but at a slower rate.
-
Deficiency of C1INH (HAE): This leads to recurrent episodes of angioedema. See Angioedema (Hereditary).
-
Mannose-binding lectin (MBL) pathway deficiency
- Newborns with MBL deficiency are at increased risk for infection. Individuals with MBL deficiency have a higher propensity for severe infection in early life.
- In young children and newborns with repeated infections, MBL defects should be considered.
- Worsening clinical features are also reported in patients with rheumatoid arthritis and MBL deficiency.
- In a study of patients with cystic fibrosis (CF) with or without MBL variant alleles, Garred et al found the following: [3]
- Lung function in carriers of MBL variant alleles was significantly reduced compared with patients with normal homozygotes.
- The negative impact of variant alleles on lung function was especially confined to patients with CF and chronic Pseudomonas aeruginosainfection.
- Burkholderia cepacia (previously named Pseudomonas cepacia) infection was significantly more frequent in patients with CF and MBL variant alleles.
- Using a modified life table analysis, the authors estimated that the predicted age of survival was reduced by 8 years in patients with CF who carried variant alleles compared with normal homozygotes with CF.
-
C3 nephritic factor (C3NeF) and mesangiocapillary glomerulonephritis (MCGN)
- C3Nef is an autoantibody that binds to and stabilizes the C3 convertase (C3bBb).
- The association of C3NeF and MCGN, especially MCGN type II, has been well defined. Different isotypes of C3NeF are recognized, the main one being an IgG autoantibody against factor H. Factor H is an important regulator of the C3 conversion step in the alternate pathway. C3NeF inhibits functions of factor H, which leads to overwhelming complement activation at the stage of C3 conversion. Continuous C3 activation in vivo results in the well-known consequences of very low serum levels of C3 in MCGN.
- C3NeF may act directly within glomeruli to cause local complement activation and ensuing renal damage.
- C3Nef is also associated with partial lipodystrophy.
-
Factor H deficiency
- Factor H deficiency is associated with hemolytic-uremic syndrome (HUS), especially in familial cases that involve homozygous factor H deficiency.
- Factor H deficiency also occurs with membranoproliferative glomerulonephritis.
- Parents have one half the normal levels of factor H.
- HUS associated with factor H deficiency is characterized by verotoxin-negative (diarrhea-negative) HUS. Atypical and recurrent HUS is also seen in CD46 deficiency.
-
-
Causes
See the list below:
-
Classical pathway deficiencies (C1, C4, C2) are inherited in an autosomal recessive pattern.
-
C3 deficiency is inherited in an autosomal recessive pattern.
-
No known homozygous deficiency of factor B has been reported, although one heterozygous case has been described. Properdin deficiency is the only X-linked complement deficiency. All reported cases of properdin deficiency involve males.
-
Late complement component deficiencies (ie, C5, C6, C7, C8, C9) are inherited in an autosomal recessive fashion.
-
Laboratory Studies
The following studies may be indicated in complement deficiency:
-
Measurement of CH50 is the best screening test for deficiency of classical or terminal pathways. Hemolytic activity of the complement system is measured as hemolysis of sheep erythrocytes sensitized by specific antibodies. The degree of hemolysis is measured by CH50.
- One CH50 unit is defined as the volume or dilution of serum that lyses 50% of erythrocytes in the reaction mixture.
- AH50 is an analogous test to measure alternate-pathway function. This test is available only in specialized laboratories.
-
Low levels of CH50 or AH50 warrant further evaluation. A low level of both CH50 and AH50 suggests deficiency of one of the components shared by both pathways (ie, C3-C9).
-
A low CH50 level suggests deficiency of a classical or terminal C component.
- C1-C8 deficiency: CH50 value is approximately 0.
- C9 deficiency: CH50 value is approximately one half of the normal value.
-
A low AH50 level suggests a deficiency in factor B, factor D, or properdin.
-
Immunochemical methods are used to demonstrate specific complement protein deficiencies by using specific antibodies for each component. In certain cases, functional properties of complement components are diminished despite the presence of normal amounts of protein component detected by immunochemical measures. Functional assays are required for further evaluation.
-
Acquired deficiencies vary with the severity of the underlying disorder.
- Low C3 and C4 levels suggest activation of the classical pathway.
- Low C3 and normal C4 levels suggest activation of the alternate pathway.
- ANA is generally negative in immune complex diseases caused by complement deficiency.
-
Hereditary angioedema (HAE) may be reflected as the following:
- Low levels of C4 but normal levels of C3
- C1 inhibitor (C1INH) – Low levels in type I (quantitative deficiency of C1INH) and may be normal in type II (functional deficiency of C1INH)
-
Function of the mannose-binding lectin (MBL) pathway can be assessed with enzyme-linked immunosorbent assay (ELISA), which detects C4d bound to mannan that is coated to the plate after the test serum was incubated in the plate. Serum levels of MBL and MASP-1 and MASP-2 can be measured immunochemically, and some commercial laboratories offer such measurement techniques. MBL deficiency can be analyzed genetically (single nucleotide polymorphisms [SNPs] of exons of MBL) only in specialized laboratories.
-
Plasma levels of regulatory proteins such as factor H and factor I can be measured in specialized laboratories.
Complement Deficiency Treatment & Management
Medical Care
See the list below:
-
No specific treatment is available for genetically acquired complement deficiencies; however, acute attacks of hereditary angioedema (HAE), C1INH deficiency, have been successfully treated with infusion of vapor-heated C1 esterase inhibitor. Androgen therapy can be used to prevent HAE attacks. These treatments are recommended only in adults. A study in the Netherlands indicated efficacy of self-administration of plasma-derived C1INH concentrate for prevention and treatment of angioedema attacks in patients with C1INH deficiency.
-
Only supportive therapy is available for other complement deficiencies. Fresh frozen plasma is used for emergent replacement of complement components.
-
Genes have been cloned for individual component deficiencies. Therefore, gene therapy may be a choice in the future.
-
All routine vaccines are recommended in complement deficiency.
- Meningococcal vaccine is recommended for children with early or terminal complement component or properdin deficiencies.
- Pneumococcal vaccine is recommended for deficiency of early components. The effects of influenza plus pneumococcal conjugate vaccination in preventing respiratory tract infections was recently studied.
***************************************************************
Complement Deficiencies
Complement is the term used to describe a group of serum proteins that are critically important in our defense against infection. There are deficiencies of each of the individual components of complement. Patients with complement deficiencies encounter clinical problems that depend on the role of the specific complement protein in normal function.
Description of the Complement System and Its Pathways
The complement system consists of more than 30 proteins, present in blood and tissues, as well as other proteins anchored on the surfaces of cells. The primary functions of the complement system are to protect from infection, to remove particulate substances, (like damaged or dying cells, microbes or immune complexes) and to help modulate adaptive immune responses.
As part of the innate immune system, complement acts immediately to start the process of removal and resolution of the problem. Complement works with the inflammatory cells of the innate immune system and those of adaptive or acquired immunity. It also interacts with proteins of the coagulation and kinin generating systems along with others.
Complement activation is tightly regulated and designed to kill invading microbes while producing minimal “collateral damage” that could result in the destruction of host tissues. Complement proteins in the circulation are not activated until triggered by an encounter with a bacterial cell, a virus, an immune complex, damaged tissue or other substance not usually present in the body.
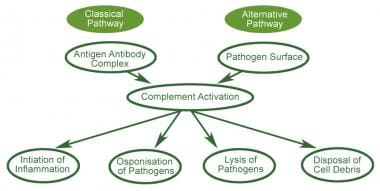
Complement activation is a cascading event like the falling of a row of dominoes. It must follow a specific order if the end result is to be achieved. The circulating proteins have been grouped into three activation pathways, based on the types of substances and proteins that initiate the activation.
If you visualize a trident, the three tines represent the different initiation routes, while the handle represents the lytic mechanism by which this cascade ultimately destroys the threat, no matter which activation pathway started the response. The diagram in Figure 1depicts the activation pathways.

-
-
The Classical Pathway (CP) is activated primarily by immunoglobulins (antibodies, including autoantibodies) that are bound to antigens – either in the fluid phase as soluble immune complexes, or on cell membrane surfaces or other tissues. Aggregates of immunoglobulins such as cryoglobulins also activate the CP. Components of the CP are C1q, C1r, C1s, C2 and C4. The CP was the first to be discovered, but is the most recent in evolutionary terms.
The Lectin Pathway (LP) is similar to the CP except for the first two steps. Mannose binding lectin (MBL), the Ficolins, and Collectin can initiate the LP. Associated with these are enzymes referred to as MASPs (MBL-Associated Serine Proteases). C2 and C4 also participate in the LP. The LP is thought to be the most evolutionarily primitive of the complement pathways and the first to react before the adaptive immune response occurs.
The Alternative Pathway (AP) is initiated by fragments of the complement component C3. Other elements of the AP are Factor B, Factor D and properdin. A unique feature of the AP is the presence of the only positive regulator in the complement system, Properdin.
Properdin makes it possible for the amplification loop of the alternative pathway to set up a very efficient mechanism for putting lots of C3b onto the surface of the activating cells, protein complexes or particles in the immediate vicinity of the activation site.
Because the ability of the C3b to bind to these surfaces decays rapidly, the activation is limited to just the region around the C3 cleavage site. This time-limitation is another control mechanism for the complement pathway.
The Terminal Pathway (TP) is the final set of steps in the complement activation process that forms a membrane lesion or hole (membrane attack complex or MAC) that kills susceptible bacteria or other cells that activate complement on their surfaces.
The TP is dependent upon at least one of the other pathways to initiate the process that it then completes. The components of the TP are C3, C5, C6, C7, C8 and C9. A fluid phase form of the MAC, called the Terminal Complement Complex (TCC) can be found in the circulation after complement activation occurs and makes a useful laboratory marker for complement activation.
Control mechanisms to prevent unregulated activity (and tissue damage) are present in each pathway. C1-esterase inhibitor (C1-inh) is a serine protease inhibitor (SERPIN) that acts by forming a complex with active enzymes to trap and inactivate them. It is important in controlling the C1r and C1s activation in the CP, and the MASPs in the LP along with several enzymes in the coagulation system.
The dynamic interplay among the different complement pathways and their control processes involves other plasma protein systems such as enzymes of the coagulation system, enzymes from inflammatory cells, and substances such as histamine released from cells in the local environment. All of these participants affect the outcome of an activation event.
Most of the time, the outcome is favorable to the host, with the danger met and the situation returned rapidly to normal. The diseases that accompany uncontrolled activation or inadequate performance of complement’s functions are often the result of inherited deficiency or subtle impairment of one or more of the components.
Complement Deficiencies and Their Diagnosis
Clinical indications for possible complement deficiencies include recurrent mild or serious bacterial infections, autoimmune disease, or episodes of angioedema (a painless, but often dramatic, swelling under the skin, or swelling in the intestines, which can be extremely painful). Very rarely angioedema in the brain can be fatal. This swelling does not respond to antihistamines or epinephrine.
The list of potential complement-related problems includes renal disease, vasculitis (blood vessel inflammation) and age-related macular degeneration. A history of family members having the same presentation should increase the suspicion of an inherited complement deficiency, most of which are inherited as autosomal co-dominant conditions.
All genes, except for those in the Male sex chromosome Y, come in pairs, one inherited from mom and one from dad. Co-dominance occurs when the contributions of both alleles are visible in the phenotype. In the ABO blood group example, the A and B allele classes are co-dominant in producing the AB blood group phenotype, in which both A-type and B-type antigens are made.
By contrast, with traditional dominant–recessive gene combination like eye color, a single brown allele is dominant and if the other parent contributed a blue color allele, the eyes will be brown rather than a mix of brown and blue. In this context it means both the normal and mutant complement proteins are produced in the affected individuals.
There is an exception in the case of Properdin, the gene for which is on the X chromosome and is inherited as an x-linked disease. (See chapter titled “Inheritance.”) The initial tests done to evaluate a patient’s complement system are critical because they can often identify an inherited defect and indicate what further testing must be done to make the diagnosis.
The aim of the evaluation process is to clearly define the complement component deficiency with as few tests as possible, while ruling out acquired causes of low complement values. Several screening tests are available that make it easier to find the answers. It is important to know as much as possible about the reason(s) for low or absent complement so that decisions regarding appropriate treatment can be made, including when to use antibiotics and immunizations as well as genetic counseling for inherited deficiencies.
Therapeutics specific for complement deficiencies are still in the developmental stage for most components, but in some cases, such as C1-Inh deficiency, there are currently several drugs available. For uncontrolled complement activation as in PNH or due to dysfunctional FH, there are a few drugs available to treat acute episodes or to prevent recurrence.
Therapeutics for complement-derived diseases is in its infancy at this time, but more treatments should become available in the near future.
Deficiencies in the Classical Pathway: C1q, C1r, C1s, C4, C2, C1-Inh
Rapid clearance of immune complexes, dying cells and debris from damaged tissues is a job that is performed efficiently by a normal CP. Primary deficiency of C1q, C1r, C1s or C4 is closely linked to development of systemic lupus erythematosus (SLE) or rheumatoid arthritis (RA), thought to be due in part to the inability of complement to clear immune complexes and dying cells.
Small complexes are cleared from the circulation when they bind to complement receptors on macrophages in the spleen and liver. Without complement, the complexes can grow too large to be easily cleared. The resulting aggregates can activate the alternative pathway, allowing C3 to be deposited into the matrix, with re-solubilized complexes that can be dealt with by the clearance through the liver and spleen.
Failing this, these large complexes are no longer soluble, and form deposits in the tissues and become a site of inflammation. Dying cells, if not cleared by non-inflammatory CP activity, may serve as sources of altered self-antigens with the potential for inducing autoantibodies.
C2 deficiency is the most common complement deficiency in Caucasian populations, with frequency estimates between 1 in 10,000 to 1 in 20,000 for homozygous C2-deficient patients. C2 deficiency is found in a slightly higher proportion of SLE patients compared to healthy controls.
In primary immunodeficiency, C2 deficiency is found in young children who have recurrent infections, primarily upper respiratory infections with Streptococcus pneumoniae or similar organisms. These children often have frequent ear infections and colds.
Hereditary angioedema (HAE) is a disease caused by deficiency of the CP control protein, C1-Inh. Symptoms generally begin around puberty but can occur earlier. These individuals have recurrent swelling in the extremities, face, lips, larynx or GI tract.
The patients describe a sensation of fullness but not pain or itching in the affected area except for those with abdominal swellings who often experience acute abdominal pain. The latter two presentations are of the most concern because suffocation can occur if the airways are obstructed, and the acute swelling of the abdominal region produces intense pain often resulting in exploratory surgery.
The mechanism for production of the swelling involves not the complement enzymes, but the kinin-generating pathway. It is the production of Bradykinin through this pathway that is responsible for the tissue permeability changes that cause the swelling.
Acute treatments include C1 inhibitor, a replacement therapy (both plasma derived and recombinant products are available); ecallantide, a kallikrein inhibitor; and icatibant, a bradykinin-2 receptor antagonist. Prophylactic treatments include attenuated androgens and C1 inhibitor.
Deficiencies of the Lectin Pathway Components
MBL, M-ficolin, L-ficolin, H-ficolin, CL-11, MASPs
Manose Binding Lectin Deficiency is Not a Primary Immunodeficiency Disease.
MBL (manose binding lectin) is a part of the lectin pathway of the complement system, one of several different components of our immune defense. The lectin pathway may be first to react before a traditional immune response occurs. It was thought that deficiency of MBL might explain some cases of increased susceptibility to bacterial infection.
However, when a test was developed to measure MBL in the blood, it was determined that low or absent MBL is very common, affecting approximately 5-30% of all individuals. Therefore, its absence alone cannot be a cause of serious immunodeficiency or a large portion of the world’s population would suffer from frequent major recurrent and potentially fatal infections.
Unfortunately, the MBL test is occasionally still ordered during an evaluation for immunodeficiency and, when the results show MBL to be low or absent, it is wrongly interpreted as indicating the presence of a primary immunodeficiency disorder. By contrast, expert immunologists experienced in caring for PI patients believe that low or absent components of this lectin system, including low or absent MBL, do not cause immunodeficiency by themselves.
There is no recommended treatment for low or absent MBL, and immunoglobulin replacement therapy is clearly not indicated for that purpose.
It is important to stress that the finding of low or absent MBL does not indicate that the cause for an individual’s infections has been found and that the diagnostic process must continue until the correct diagnosis is determined.
In such cases the IDF recommends that referral to an experienced immunologist to assist in the diagnostic evaluation be considered.
Deficiencies of the Alternative Pathway
Factors D, B and Properdin
Factor D deficiency is very rare and has only been described in two families. Both of these families had multiple members with a history of serious infections. Factor B is an acute phase protein and increases during inflammation.
There is only one unconfirmed report of this deficiency in humans. Properdin is the only complement protein that is X-linked. The protein is synthesized by monocytes, granulocytic cells and T-cells. Several mutant forms of the protein have been identified that result in decreased AP function.
Properdin deficiency increases the susceptibility to bacterial infections of the Neisseria family of organisms. The most prominent in the group is N. Meningitis, the cause of a serious form of meningitis. Typical family histories include male relatives who have had or died from Neisserial infections.
Alternative Pathway Control Proteins
Deficiencies of factor H are linked with a wide variety of symptoms. Complete deficiency of H leads to uncontrolled activation of the AP and depletion of C3 occurs.
This form of factor H deficiency is similar in presentation to the late component deficiencies due to the low or absent levels of C3. Recent data has been published that demonstrates how critical the role for this complement control protein is in maintaining health in a number of tissues.
In addition to bacterial infections, deficiency or dysfunction of factor H and the resulting dysregulation of the AP is associated with various forms of kidney disease including atypical Hemolytic Uremic Syndrome (aHUS), as well as age-related macular degeneration (AMD). These diseases are examples of control processes gone awry on the surfaces of the organs affected.
Treatment of Complement Deficiencies
Deficiencies of the early classical and lectin pathway components are primarily accompanied by upper respiratory infections, otitis media, along with lupus-like symptoms.
Any complement deficiency should be treated as an immune deficiency, and the patient should be immunized against the likely candidate microbes for their deficiency. Antibody responses should be checked after vaccination, since the inability to activate complement impairs the immune response to some extent.
Currently, there are no specific treatments for complement deficiencies. Infection prevention and appropriate treatment of infections (usually with antibiotics), when they do occur is key in the care of patients with these deficiencies.
Fresh frozen plasma has been tried in some cases, but carries the risk that the patient may make antibody to the missing complement component, so prolonged use is not advised. Prophylactic antibiotics can be used if the patient experiences repeated infections, and increased vigilance with rapid treatment of problems is another option. Most of these patients eventually make antibodies against the offending bacteria and do not get sick as often.
Boys with Properdin deficiency (X-linked) should be immunized against Neisseria meningitidis, in addition to the usual vaccinations of childhood. Often there is a family history of an uncle or other relative who died from Neisserial infection at an early age.
Deficiencies of the other alternative pathway components and the terminal pathway proteins are also susceptible to Neisseria meningitidis and should be immunized. The vaccine titers should be verified in these individuals as well.
****************************************************
Complement Deficiency
Background
The complement system is a part of the immune system that is concerned with the innate immunity. It consists of numerous proteins that are activated by several triggering factors and start a chain of events in the human body. Because the pathway finally culminates in the creation of a cytolytic “membrane attack complex”, it is a vital mechanism to fight infection.
In 1896, Jules Bordet, working at the Pasteur Institute in Paris, demonstrated that the factor in the blood serum capable of killing bacteria could be analyzed into two components: heat stable and heat labile. The heat labile component is what is now known as the “complement.” The term complement was introduced by Ehrlich in the late 1890s.
In the 1900s, the complement system was understood to be a cytolytic system that primarily lysed bacteria and erythrocytes, which were sensitized with antibody. The complement system is currently known to contain at least 30 different proteins that are primarily formed in the liver and circulate in their inactive form. When activated, these proteins produce various complexes that play a major role in the innate and adaptive immune defense.
The 3 major pathways of complement activation are as follows:
- The classical pathway (termed classical because it has been studied for >100 y and was the first pathway to be discovered)
- The alternate pathway
- The mannose-binding lectin (MBL) pathway
Deficiency of each component of the classical and alternate pathways is rare and comprises less than 1% of patients with primary immunodeficiency. Deficiency of components of the MBL pathway appears to be fairly common. Deficiency of early components of classical pathways has been associated with autoimmune disease, whereas deficiency of late components of complements lead to increased susceptibility to certain infections.
See the image below.
 Scheme showing the cascade of events during the activation of the complement system.
Scheme showing the cascade of events during the activation of the complement system.Nine complement components in the classical pathway are designated by a capital letter C and numbers 1-9. Two proteins that participate in the alternate pathway are termed factors and are represented by capital letters B and D. Proteolytically cleaved components of proteins are expressed by lowercase letters (eg, C2a, C2b). Inactive components are designated with an “i” (eg, inactivated C3b is termed iC3b).
Pathophysiology
The binding of C1 to antigen-antibody complexes that contain immunoglobulin M (IgM) or immunoglobulin G (IgG) antibodies (subclasses IgG1, IgG2, and IgG3) activates the classical pathway. Only these Ig isotypes have complement-binding sites in the Fc portion (CH2 domain of IgG and CH3 domain of IgM).
C1 is a large multimeric protein complex composed of 3 subunits: C1q, C1r, and C1s. C1r and C1s are serine esterases. The C1q must bind to the 2 Fc portions of immunoglobulin (Ig) heavy chains to initiate the complement cascade. [1] The binding of C1q to the immune complex leads to enzymatic activation of C1r, which, in turn, cleaves and activates C1s. Activated C1s cleaves C4 into C4a and C4b. A single activated C1s can cleave numerous molecules of C4. This process leads to continuous formation of C4b but is inhibited by C1 esterase inhibitor enzyme (C1INH). Deficiency of C1INH leads to uninhibited formation of C4b and C4a.
C4b has an internal thioester bond that allows C4b to form covalent amide or ester linkages with the immune complex or the cells coated with antibodies. C2 then complexes with immune complex or cell surface–bound C4b and is cleaved into C2a (soluble component) and C2b by C1s. The C2b remains physically associated with C4b on the target cell surface, forming classical pathway C3 convertase (C4bC2b) that proteolytically cleaves C3 into C3a and C3b.
Once C3 is cleaved, all 3 complement activation pathways share the same terminal complement components (C5-C9). The classical pathway is important in antigen-specific adaptive immune defense because it is activated by antigen-antibody complexes. Complement activation via the classical pathway effectively lyses antibody-coated pyogenic bacteria such as Streptococcus pneumoniae and Haemophilus influenzae and cells coated with antibodies (often microbe-infected cells).
Mannose-binding lectin pathway
MBL is an acute-phase protein. It is secreted by hepatocytes as a part of the innate immune defense and activates complement cascade without antigen-antibody complex.
The MBL binds to mannose residues of polysaccharide on the microbial surface. The MBL-associated serine proteinases MASP-1 and MASP-2, analogous to C1r and C1s, cleave C2 and C4, forming C4b/C2b C3 convertase. The terminal components of complement are then activated. The MASP-1 can also directly cleave C3. The MBL has high structural homology with C1q protein and can bind with C1r and C1s, with subsequent activation of C4 and C2. The MBL pathway is generally thought to provide initial antigen-independent immune defense by rapid activation of complement cascade, directly recognizing sugar residues of microbes.
Alternative complement pathway – Properdin, factor B, and factor D
This pathway is activated by the combination of factor B, factor D, and properdin.
Properdin was first described by Pillemer and colleagues, and its main function is to stabilize the alternative pathway convertase. It is also referred to as factor P. Factor B is a single polypeptide chain glycoprotein with a molecular mass of 93 kDa and is the zymogen of the alternate pathway C3/C5 convertase. Factor D is a 24-kDa single-chain serine proteinase. It is present in the concentration of 1.4-2.2 mg/L, but the levels are increased ten-fold in patients with renal failure. Large amounts are found in the urine of patients with proteinuria, in particular those suffering from Fanconi syndrome.
C3 in the plasma is continuously cleaved at a low rate (C3 tick over). If active C3 (C3b) attaches to the cell surface that lacks complement regulators, it permits rapid amplification of the complement cascade. Namely, factor B binds to C3b bound to the cell surface by forming amide or ester bonds. Factor B is then cleaved by factor D to generate Bb, which forms alternate pathway C3 convertase (C3bBb). Properdin stabilizes the C3bBb complex.
After the C3 convertase cleaves another C3 bound to the convertase, C3b combines with C3 convertase complex to form the alternate pathway C5 convertase. The C5 convertase further activates C5 to C5b, and sequential binding of terminal components C6, C7, C8, and C9 forms the membrane attack complex (MAC). The alternate pathway, along with the MBL pathway, plays an important role in innate immune defense in an antigen-independent manner. Moreover, even when C3b is generated by the other two pathways, it can form a complex with Bb that can further cleave more C3. Thus, the alternative pathway C3 convertase also amplifies complement activation initiated by other pathways.
C3 component
The central event in complement activation is proteolysis of C3, which produces C3b and C3a. As described above, C3b that is covalently attached to the cell surface or antigen-antibody complex initiates late steps of complement activation common to all 3 pathways, leading to the formation of lipid-soluble pore structures of the MAC. Along with C5a and C4a, C3a functions as an anaphylatoxin, inducing degranulation of mast cells and basophils. They also function as chemotactic factors for leukocytes. C3b is cleaved into C3d, C3dg, and iC3b (inactive C3b) by factor I. C3b and C3b cleavage products bind to complement receptors expressed on various cells and initiate other immune responses. Membrane-bound C3b and iC3b act as opsonins by binding receptors on neutrophils and macrophages. C3d and C3dg augment B-cell responses to antigen.
Late complement components – C5-C9
Converting C5 to C5a and C5b marks the activation of the terminal complement components. These events, in turn, lead to the formation of MAC by C5, C6, C7, C8, and C9. The MAC has a hydrophilic center that increases leakage of water and ions through the cell membrane, causing osmotic lysis and cell death.
Regulatory proteins
These are the proteins that play vital roles in different steps of the cascade reactions of the complement pathways. Some of the important proteins, along with their functions, are as follows:
-
C1INH: This is an inhibitory protein that regulates the classical pathway by covalently attaching to C1r2-C1s2, which dissociates C1r2-C1s2 tetramer from C1q and stops activation of the classical pathway.
-
C4BP: This regulates the classical pathway by dissembling the C4bC2b complex. C4BP binds to C4b and blocks binding of C2b. Decay-accelerating factor (DAF) and complement receptor 1 (CR1) also bind to C4b to block C4b/C2b complex formation. DAF also displaces Bb from C3b.
-
Factor I: Factor I regulates the complement activation by cleaving C3b. Factor I cleaves cell membrane–associated C3b into iC3b, C3d, and C3dg. It also cleaves C4b. CD46, factor H, C4BP, and CR1 all serve as cofactors for factor I–mediated cleavage of C3b/C4b. [2]
-
Factor H: Factor H promotes conversion of C3b to iC3b with factor I and displaces Bb from the alternate pathway C3 convertase (C3bBb). Cell membrane rich in sialic acid has higher binding affinity to factor H than to factor B. Because mammalian cells have higher sialic acid content than microbial cells, factor H prevents excessive complement activation in host cells but not in microbes. Factor H is the major regulator in the alternate pathway activation.
-
CD59: CD59 expressed by many cell types inhibits binding of C9 to the membrane-inserted C5b-8 complex and limits MAC formation on host cells. CD59 is not expressed by microbial cells, which allow complement-mediated microbial cell lysis, sparing host cells.
-
Membrane cofactor protein: This is a cofactor for factor I–mediated cleavage of C3b and C4b.
-
DAF: This accelerates decay of C3/C5 convertase.
-
CR1: This is the receptor for C3b/C4b and has an inhibitory profile similar to MCP and DAF.
-
Properdin: This stabilizes AP convertases.
-
Clusterin: This blocks fluid phase MAC.
-
S protein: This blocks fluid phase MAC.
-
Anaphylatoxin inactivator: This inactivates C3a, C4a, and C5a.
-
Complement system: This is an important regulator of B-cell activation.
Naive B cells receive a stimulus from the B-cell receptor (BCR), leading to activation, elimination, or anergy. The strength of signal transduction of B cells and other antigen-presenting cells (eg, dendritic cells) depends not only on the affinity of antigen binding but also on positive signals via the B-cell coreceptor complex composed of CD21 (complement receptor 2 [CR2])/CD19/CD81(TAPA-1). CD21 binds to antigens via attached C3d while membrane Ig binds to the antigen. This allows an association of CD19 to BCR-associated kinases and rapid phosphorylation of the cytoplasmic tail of CD19, leading to activation of PI-3K, which, in turn, augments BCR-initiated B-cell activation signaling. Thus, the absence of CD21 expression by B cells leads to impaired humoral immune response to T-dependent antigens. This is characterized by a decrease in B-cell follicular retention and germinal center survival.
C3b cleavage products have also been shown to augment immune memory; C3 receptors expressed on dendritic cells bind to both immune complexes and antigen alone. CD21 (CR2) expressed on follicular dendritic cells serves to trap iC3b-coated and C3dg-coated antigen-antibody complex in the germinal center.
Complement deficiency
Deficiencies of the complement components have been reported for most of the constituents. These deficiencies can be inherited or acquired and complete or partial.
Complement deficiency and disease association includes the following:
-
Deficiency of early components of classical pathway (C1, C4, C2): Autoimmune disease, especially systemic lupus erythematosus (SLE), is the most common presentation in patients with early component deficiency. The incidence rates of SLE in individuals with C1q and C4 are reported to be around 90% and 75%, respectively. Patients with C2 deficiency develop SLE with lesser frequency (around 15%). The proposed mechanisms of high incidence of autoimmune diseases include impaired clearance of immune complex and apoptotic cells and loss of complement-dependent B-cell tolerance. Recurrent bacterial infection is common in patients with C2 deficiency. Atherosclerosis also appears to occur at a higher frequency in individuals with C2 deficiency.
-
MBL pathway
- MBL deficiency is linked with frequent pyogenic infection, including pneumococcal infection in infants and young children. Severe pneumococcal disease is also reported in patients with MASP-2 deficiency.
- In addition, MBL deficiency is 2-3 times as common in patients with SLE as in the general population. Others also report an increase in cardiovascular diseases associated with atherosclerosis in patients with MBL deficiency.
- Schizophrenia is a severe mental disorder, with worldwide prevalence of 1–1.5%. Immunological research in schizophrenia indicates that infectious or autoimmune processes might play a role in the etiopathogenesis.
- The complement system is a major mediator of innate immune defense against infection and contributes to many functions of the immune system including inflammation, opsonization and cell lysis.
- MBL activates the complement system via the lectin pathway.
- Inherited MBL deficiency, common in most human populations, predisposes to infectious and autoimmune diseases.
- A modulatory role for MBL in autoimmune disease has been reported. Investigations in Denmark and Hong Kong have suggested that MBL variant alleles are associated with both severity and early onset of disease in patients with rheumatoid arthritis. [3] However, patients with late-onset disease who were also homozygous for wild-type MBL genes were more likely to show evidence of persistent inflammation.
-
Alternative pathway (properdin, factor B, factor D): This is associated with severe fulminant neisserial infections with a high mortality rate.
-
C3, factor H, and factor I: Deficiency of these factors predisposes individuals to severe pyogenic bacterial infections. Factor H and factor I deficiencies cause secondary C3 deficiency with C3 consumption and impose the same infectious risk as primary C3 deficiency. Factor H deficiency is also associated with atypical (diarrhea-negative) hemolytic-uremic syndrome (HUS) and glomerulonephritis. C3 deficiency is associated with membranoproliferative glomerulonephritis.
-
Terminal pathway (C5-C9): The lack of MAC formation results in severe recurrent infection by Neisseria gonorrhoeae or Neisseria meningitidis.
-
C1INH: Primary and secondary deficiency of C1INH leads to uninhibited cleavage of C4 by C1s. Hereditary angioedema (HAE) is caused by heterozygous deficiency of C1INH and characterized by recurrent episodes of angioedema, which usually subside within 48-72 hours. C1INH is a regulator for C1r/C1s, as well as for Hageman factor, clotting factor XI, plasma kallikrein, and plasmin. Angioedema is caused by various mediators that involve these pathways.
-
DAF and CD59: Defects of these 2 regulatory proteins make erythrocytes highly susceptible to complement-mediated cell lysis. This is typically seen in patients with paroxysmal nocturnal hemoglobinuria (PNH), which has clinical features that are characterized by hemoglobinuria and venous thrombosis of major vessels.
History
The following may be associated with complement deficiency:
-
Patient or family history of recurrent systemic infection caused by encapsulated bacteria, especially meningococci
-
Family history of fulminant meningococcal disease occurring in males, suggestive of X-linked properdin deficiency
-
Meningococcal disease occurring in persons older than 10 years, especially when caused by non–group B meningococci (especially serogroups Y and W-135)
-
Family history of systemic lupus erythematosus (SLE) or occurrence of atypical features of SLE, especially negative lupus serology findings (negative antinuclear antibody [ANA] and negative dsDNA findings)
-
Specific syndromes such as partial lipodystrophy, angioedema, and paroxysmal nocturnal hemoglobinuria (PNH): Consider evaluation of the complement system in patients with these syndromes.
-
Recurrent pyogenic infection, including severe pneumococcal diseases in infants and young children with normal humoral immune workup findings
Physical
See the list below:
-
Classical pathway deficiencies – C1, C4, and C2
- Deficiencies of the classical complement pathway have been strongly linked to the development of autoimmune disorders, especially those in which excessive immune complexes are formed. Patients may develop collagen vascular disorders, mainly SLE. Ninety-five percent of patients with C1q deficiency, 75% of those with C4 deficiency, 30% of those who are deficient in C2, and 50-55% of those who are deficient in C1r and C1s (deficiency usually involves both C1r and C1s) develop collagen vascular diseases (mainly SLE). Evidence suggests that this may result from the combination of impaired complement-dependent B-cell tolerance and impaired clearance of immune complex and apoptotic cells.
- C1 deficiency generally leads to severe immune complex disease with features of SLE and glomerulonephritis. Serum levels of anti-C1q antibodies were increased significantly in patients with SLE and proliferative nephritis. Among different serological parameters for assessing the renal activity of SLE, only anti-C1q antibodies titers showed significant differences between quiescent and active lupus nephritis. Patients with C2 deficiency are reported to reveal anti-C1q antibody in the absence of anti-dsDNA.
- Complete C4 deficiency is rare. Two genes, C4A and C4B, encode the C4 complement. Both genes are highly polymorphic, and, to date, at least 35 different alleles have been described. Almost all patients with complete C4 deficiency have discoid or SLE, with or without associated glomerulonephritis.
- C2 deficiency has been the most commonly reported classical pathway defect. Skin and joint manifestations are common, and renal disease is relatively rare. Patients with C2 deficiency are also reported to have recurrent or invasive infections.
-
C3 deficiency
- C3 deficiency is rare. Because of its importance as a convergence point of the 3 complement pathways, all patients with C3 deficiency develop recurrent, severe, pyrogenic infections early in life. Some patients may also develop membranoproliferative glomerulonephritis.
- C3 deficiency leads to an inability to formulate membrane attack complex (MAC), thus markedly compromising chemotactic and bactericidal activities of the complement cascade.
- Patients with C3 deficiency generally have less than 1% of the normal amount of C3 antigen and C3 function in their serum.
- Because of ineffective opsonization of pathogens, patients with C3 deficiency present with severe recurrent pyogenic infections, mainly caused by meningococci and pneumococci.
- According to published studies, 78% of patients have repeated infections, and 79% experience autoimmune disorders, such as arthralgia and vasculitic rashes, lupuslike syndrome, and membranoproliferative glomerulonephritis.
-
Alternate pathway deficiencies (properdin, factor B, factor D)
- These deficiencies are rare. Properdin deficiency is associated with increased risk of infections with encapsulated bacterial organisms. Patients often present with a history of invasive meningococcal diseases, as well as severe pneumococcal and H influenzae infection. Recurrent infections may be rare in patients with properdin deficiency because the classical pathway can take over the actions of the alternate pathway once antigen-specific adaptive immune responses are established with immunoglobulin (Ig)G and IgM antibody formation.
- Only 3 adults with factor D deficiency have been described in the literature. A 6-day-old newborn boy with pneumococcal sepsis and meningitis was diagnosed with complete deficiency of factor D and diminished functional factor B. The addition of purified factor D restored the capacity of the patient’s serum to generate the alternate pathway fluid-phase C3 convertase (C3bBbP) in response to zymosan. This also restored activity of factor B in the patient’s serum. In contrast, the addition of factor B did not alter factor D activity in the serum, indicating requirement of factor D for function of factor B.
-
Late complement component deficiencies – C5, C6, C7, C8, and C9
- MAC cannot be formed; hence, bactericidal activity is depressed.
- Patients are susceptible to recurrent pyogenic infections.
- Patients typically present with meningococcal meningitis or extragenital gonococcal infection.
- Two thirds of patients experience at least one episode of meningococcal disease, and as many as one half of patients experience recurrent neisserial infections.
- Patients with C9 deficiency have the ability to kill Neisseria, but at a slower rate.
-
Deficiency of C1INH (HAE): This leads to recurrent episodes of angioedema. See Angioedema (Hereditary).
-
Mannose-binding lectin (MBL) pathway deficiency
- Newborns with MBL deficiency are at increased risk for infection. Individuals with MBL deficiency have a higher propensity for severe infection in early life.
- In young children and newborns with repeated infections, MBL defects should be considered.
- Worsening clinical features are also reported in patients with rheumatoid arthritis and MBL deficiency.
- In a study of patients with cystic fibrosis (CF) with or without MBL variant alleles, Garred et al found the following: [3]
- Lung function in carriers of MBL variant alleles was significantly reduced compared with patients with normal homozygotes.
- The negative impact of variant alleles on lung function was especially confined to patients with CF and chronic Pseudomonas aeruginosainfection.
- Burkholderia cepacia (previously named Pseudomonas cepacia) infection was significantly more frequent in patients with CF and MBL variant alleles.
- Using a modified life table analysis, the authors estimated that the predicted age of survival was reduced by 8 years in patients with CF who carried variant alleles compared with normal homozygotes with CF.
-
C3 nephritic factor (C3NeF) and mesangiocapillary glomerulonephritis (MCGN)
- C3Nef is an autoantibody that binds to and stabilizes the C3 convertase (C3bBb).
- The association of C3NeF and MCGN, especially MCGN type II, has been well defined. Different isotypes of C3NeF are recognized, the main one being an IgG autoantibody against factor H. Factor H is an important regulator of the C3 conversion step in the alternate pathway. C3NeF inhibits functions of factor H, which leads to overwhelming complement activation at the stage of C3 conversion. Continuous C3 activation in vivo results in the well-known consequences of very low serum levels of C3 in MCGN.
- C3NeF may act directly within glomeruli to cause local complement activation and ensuing renal damage.
- C3Nef is also associated with partial lipodystrophy.
-
Factor H deficiency
- Factor H deficiency is associated with hemolytic-uremic syndrome (HUS), especially in familial cases that involve homozygous factor H deficiency.
- Factor H deficiency also occurs with membranoproliferative glomerulonephritis.
- Parents have one half the normal levels of factor H.
- HUS associated with factor H deficiency is characterized by verotoxin-negative (diarrhea-negative) HUS. Atypical and recurrent HUS is also seen in CD46 deficiency.
Causes
See the list below:
-
Classical pathway deficiencies (C1, C4, C2) are inherited in an autosomal recessive pattern.
-
C3 deficiency is inherited in an autosomal recessive pattern.
-
No known homozygous deficiency of factor B has been reported, although one heterozygous case has been described. Properdin deficiency is the only X-linked complement deficiency. All reported cases of properdin deficiency involve males.
-
Late complement component deficiencies (ie, C5, C6, C7, C8, C9) are inherited in an autosomal recessive fashion.
Medical Care
See the list below:
-
No specific treatment is available for genetically acquired complement deficiencies; however, acute attacks of hereditary angioedema (HAE), C1INH deficiency, have been successfully treated with infusion of vapor-heated C1 esterase inhibitor. Androgen therapy can be used to prevent HAE attacks. These treatments are recommended only in adults. A study in the Netherlands indicated efficacy of self-administration of plasma-derived C1INH concentrate for prevention and treatment of angioedema attacks in patients with C1INH deficiency.
-
Only supportive therapy is available for other complement deficiencies. Fresh frozen plasma is used for emergent replacement of complement components.
-
Genes have been cloned for individual component deficiencies. Therefore, gene therapy may be a choice in the future.
-
All routine vaccines are recommended in complement deficiency.
- Meningococcal vaccine is recommended for children with early or terminal complement component or properdin deficiencies.
- Pneumococcal vaccine is recommended for deficiency of early components. The effects of influenza plus pneumococcal conjugate vaccination in preventing respiratory tract infections was recently studied.
-
-
-
-
